-
从头测序reads

-
青莲百奥small RNA测序

组学技术路线注 测序策略:SE50;建议数据量:一般测序10M reads,深度测序20M reads 样本要求总量 ≥ 2 μg,浓度 ≥ 50 ng/μL; OD260/280 ≥ 2.0 RIN
-
HT Barcode Sequencing of DNA, >100M Reads (screening done with Cellecta Library)

美国Cellecta公司成立于2006年4月,是一家以小核酸为平台进行药靶筛选的技术服务公司,美国众多知名药厂为其客户。Biomics Biotech作为其在中国独家代理,我们将一如既往地为您提供
-
Illumina mRNA测序数据分析结果
1. 数据预处理测序得到Raw Reads中可能含有总体质量较低、含有测序引物、末端质量偏低等不合格的Reads,这些不合格的Reads很有可能对分析质量造成一定的影响,所以必须对其进行过滤,得到
-
10× Genomics de novo基因组研究

-
转录组测序
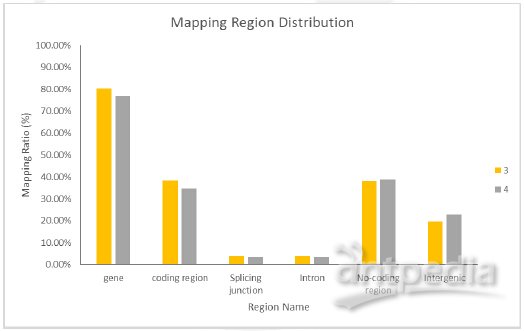
2.比对信息统计 3.随机性实验检验图 4.基因覆盖度、深度统计 这项分析主要给出每个基因被Reads 覆盖的百分比(Coverage)、碱基的平均测序深度
-
染色质免疫共沉淀测序服务

(Bridge PCR)成簇扩增4. 高通量测序5. 数据分析 A. 基本数据分析 * 原始数据读取(Raw Data, Total Reads) * 有效数据获取(Clean
-
上海伯豪外显子组测序数据分析方案

一、数据分析流程 分析流程 1.实验部分: 通过杂交富集外显子区域序列,建库,测序。, 2.序列QC: 去除低质量reads,和连续的低质量片段,去掉接头序列。QC统计reads数量及测序质量
-
App NGS RNA结合蛋白富集测序 RIP-Seq/CLIP-Seq服务

提供给客户专业的结题报告:将结合相关领域的现有研究成果,解析相关功能作用或分子机制我们的优势 测序服务流程 研究内容 1.原始数据处理:过滤掉低质量的reads,去除barcode和adapter序列
-
RNA-Seq全面解决方案

:基因组定位及统计过滤低质量序列,一般将质量低于20的替换为N。然后使用MAQ、bowtie等Mapping软件将序列定位到基因组上,统计Reads在基因组上的定位信息。Part02 :转录本表达









想在此推广您的产品吗?
咨询热线: 010-84839035
联系邮箱: sales@antpedia.net